新闻 – 第 17 页 – 卫材(中国)药业有限公司-pg电子app
卫材株式会社(总部:东京,ceo:内藤晴夫,“卫材”)宣布,渤健(nasdaq: biib)在2020年10月21日发布的2020年第三季度财报中表示,已向欧洲药品管理局(ema)提交阿尔茨海默症治疗药物aducanumab的上市许可申请。该上市许可申请须经欧洲药品管理局确认是否接受审核,渤健计划在收到通知后宣布。
媒体联络:
卫材株式会社
公共关系部
81-(0)3-3817-5120
卫材株式会社(总部:东京,ceo:内藤晴夫,“卫材”)宣布,渤健(nasdaq: biib)在2020年10月21日发布的2020年第三季度财报中表示,已向欧洲药品管理局(ema)提交阿尔茨海默症治疗药物aducanumab的上市许可申请。该上市许可申请须经欧洲药品管理局确认是否接受审核,渤健计划在收到通知后宣布。
媒体联络:
卫材株式会社
公共关系部
81-(0)3-3817-5120
卫材株式会社(总部:东京,ceo:内藤晴夫,“卫材”)近日宣布,中国国家药品监督管理局已经受理其自主研发的抗癫痫药物(aed)fycompa®(商品名:卫克泰®,通用名:吡仑帕奈)单药治疗4岁及以上患者的儿科部分性癫痫发作适应症的新药补充申请。
关于单药治疗部分性癫痫发作的申报资料是基于多项全球联合治疗临床研究(研究304、305、306和335)中评估单药治疗的安全性和有效性的亚组分析,这些研究在美国、欧洲和中国的12岁及以上部分性癫痫发作(伴或不伴继发性全面性癫痫发作)患者中进行。此外,在日本和韩国的12岁至74岁未经治疗的部分性癫痫发作(伴或不伴继发性全面性癫痫发作)患者中进行的一项iii期临床研究(freedom/研究342)的结果作为补充安全性和有效性数据提交。
关于儿科部分性癫痫发作患者的申报资料是基于一项fycompa iii期临床研究(研究311)的结果,该研究在全球范围内使用fycompa对部分性癫痫发作或原发性全面强直阵挛发作性癫痫控制不佳的儿科患者(年龄4至12岁以下)进行辅助治疗。
据估计,在中国大约有900万癫痫患者,尽管任何年龄均可能发病,但最常见的是18岁及以下儿童和青少年以及老年人。由于约30%的癫痫患者无法用目前的aeds1来控制其癫痫发作,这是一种医疗需求显著未得到满足的疾病。
fycompa是由卫材筑波研究实验室研发的一种新型抗癫痫药物,每日服用一次。该药物是一种高选择性、非竞争性的ampa受体拮抗剂,通过靶向突触后膜上ampa受体处的谷氨酸盐活性,减少与癫痫发作相关的神经元的过度兴奋。fycompa已在中国获批作为12岁及以上癫痫患者的部分性癫痫发作(伴或不伴继发性全面性癫痫发作)的辅助治疗。
卫材认为包括癫痫在内的神经学是一个重点治疗领域。随着关于fycompa的补充申请在中国获得受理,卫材继续追寻其使命,为全世界更多的癫痫患者提供有效的癫痫治疗方法。卫材致力于满足癫痫患者及其家人的各种需求,并为其提供更多福利。
媒体咨询:
卫材株式会社
公共关系部
81-(0)3-3817-5120
[编者注]
1.关于fycompa(通用名:吡仑帕奈)
fycompa是由卫材独家研发的一种新型抗癫痫药物。癫痫发作是由神经递质谷氨酸介导的,该药物是一种高选择性、非竞争性的ampa受体拮抗剂,通过靶向突触后膜上ampa受体处的谷氨酸盐活性,减少与癫痫发作相关的神经元的过度兴奋。fycompa制剂现已上市销售,每日一次,睡前口服。日本已批准片剂和细颗粒制剂。该药的口服混悬剂和片剂已在美国和欧洲获得批准。
目前,fycompa已在70多个国家和地区,包括日本、美国、中国、以及欧洲和亚洲的其他国家,作为12岁及以上癫痫患者的部分性癫痫发作(伴或不伴继发性全面性癫痫发作)的辅助治疗。此外,fycompa已被美国、日本、欧洲和亚洲等65多个国家批准作为12岁及以上癫痫患者的原发性全面强直阵挛发作性癫痫的辅助治疗。在日本和美国,fycompa获批用于单药治疗和辅助治疗4岁及以上癫痫患者的部分性癫痫发作(伴或不伴继发性全面性癫痫发作)。迄今为止,fycompa已被广泛应用于治疗全球超过300,000名患者(所有适应症合计)。卫材正在全球范围内进行一项iii期临床研究(研究338),意在将该药用于治疗与林-戈(lennox-gastaut)综合征相关的癫痫发作。此外,卫材注射制剂的开发也在进行中。
2.关于在中国提交的fycompa单药治疗部分性癫痫发作的追加申报资料所依据的iii期临床研究
中国fycompa单药治疗的额外申报资料是基于在日本、中国和韩国进行的iii期临床研究(研究3352)的结果,以及在全球(包括美国、欧洲和中国)进行的三项iii期临床研究(研究3043、3054和3065)的结果。
研究335主要评价fycompa在亚洲地区患者中的有效性和安全性。此外,研究304和305包括三个组(安慰剂、fycompa 8mg和12mg),并将评价扩展的剂量范围。研究306的关键目标是确定最小有效剂量,包括四个治疗组(安慰剂组、fycompa 2mg、4mg和8mg组)。
这些研究均为多中心、随机、双盲、安慰剂对照、平行组研究,受试者为诊断为部分性癫痫发作的12岁及以上患者,接受1至最多3种抗癫痫药物。研究335的主要终点是癫痫发作频率的百分比变化。欧洲获批的研究304、305和306的主要终点是50%缓解率(与随机化前相比,癫痫发作频率降低50%或以上的患者百分比),而在美国获批的主要终点是癫痫发作频率的百分比变化。具体来说,结果表明:
1) 研究335
2) 研究304
3) 研究305
4) 研究306
3.关于freedom(研究342)6
freedom(研究342)是一项在日本和韩国开展的评价fycompa单药治疗对12-74岁未经治疗的部分性癫痫发作伴或不伴继发性全身性癫痫发作患者的有效性和安全性的非对照、开放性、iii期临床研究。睡前口服最多4 mg fycompa,每日一次(如果发生惊厥发作,可上调至8 mg)。本研究包含一个治疗期和一个延长期,其中治疗期包括6周的滴定期和26周的维持期(如果从4 mg剂量上调至8 mg,则滴定期为4周,维持期为26周)。在本研究中,89例患者接受fycompa单药治疗,其中73例接受4 mg剂量的患者在治疗期间未出现癫痫发作,该比例超过了有效性标准*,符合主要终点。此外,中期结果表明,接受4 mg和8 mg的患者的合并比例也超过了有效性标准。本研究中观察到的最常见不良事件(发生率≥10%)为头晕、嗜睡、鼻咽炎和头痛,这与fycompa迄今为止的安全性特征一致。
*本研究中73例可评价患者的有效性标准为52.1%或更高比例的患者达到无癫痫发作,该标准主要是根据其他aed单药治疗研究的结果设定。
4.关于研究3117
研究311是一项全球性(美国、欧洲、日本、韩国)开放性iii期临床研究,评估fycompa口服混悬液作为辅助治疗药物给药时用于180名4岁至12岁以下部分性癫痫发作或原发性全身强直阵挛性癫痫发作控制不佳的儿童癫痫患者的安全性、耐受性和暴露疗效关系。这项研究包含一个治疗期(包括长达11周的滴定期和长达12周的维持期)以及一个延长期。在这项研究中,每日睡前口服一次2~16mg fycompa。主要终点是安全性和耐受性。疗效与对12岁及以上患者观察到的相似。在这项研究中观察到的最常见的不良事件(发生率为10%或更高)是嗜睡、鼻咽炎、发热、呕吐、头晕、流行性感冒、以及易怒,这与迄今为止fycompa的安全特性一致。
5.关于癫痫
癫痫在中国影响了大约900万人,在日本影响了大约100万人,在美国影响了大约340万人,在欧洲影响了大约600万人,在全世界影响了大约6000万人。由于约30%的癫痫患者无法用目前的aeds1来控制其癫痫发作,这是一种医疗需求显著未得到满足的疾病。
癫痫大致按发作类型分类,部分性癫痫发作约占癫痫病例的60%,全身性癫痫发作约占40%。在部分性癫痫发作中,异常放电干扰发生在大脑的有限区域,并可能随后扩散到整个大脑,成为全身性癫痫发作(称为继发性全身性癫痫发作)。在全身性癫痫发作中,异常放电干扰发生在整个大脑,随后可能出现意识丧失或全身出现躯体症状。
1 “癫痫和癫痫发作:研究期望。什么是癫痫?”国立神经病学与中风研究所,2016年5月24日访问,http://www.ninds.nih.gov/disorders/epilepsy/detail epilepsy.htm#230253109
2 nishida t, et al. 吡仑帕奈辅助治疗部分性癫痫发作:亚太地区随机化iii期研究。 acta neurol scand. 2018年;第137期,第392-399页。
3 french ja, et al. 吡仑帕奈辅助治疗难治性部分性癫痫发作:随机化iii期研究304。neurology. 2012年;第79期,第589-596页。
4 french ja, et al. 评估吡仑帕奈辅助治疗难治性部分性癫痫发作患者:全球随机化iii期研究305的结果。epilepsia. 2013年;第54期,第117-125页。
5 krauss gl, et al. 随机化iii期研究306:吡仑帕奈辅助治疗难治性部分性癫痫发作。neurology. 2012年;第78期,第1408-1415页。
6 yamamoto, takamichi et al. 吡仑帕奈单药治疗新诊断的局灶性癫痫发作或缓解期后的癫痫复发的有效性和安全性:开放性研究342(freedom研究)。epilepsia. 2020年;第5期,第274-284页。.
7 fogarasi, andras et al. 开放性研究-旨在研究辅助治疗药物吡仑帕奈用于儿童(4岁至<12岁)局灶性癫痫发作或全身强直阵挛性癫痫发作的安全性和有效性。epilepsia. 2020年;第61期,第125-137页。
设立社会合作项目“发现蛋白质降解药物”
东京大学(校长:makoto gonokami,“东京大学”)与卫材株式会社(总部:东京,ceo:内藤晴夫,“卫材”)近日宣布,已创建一个针对靶向蛋白质降解技术开发和新药研发的合作项目,并设立社会合作项目“发现蛋白质降解药物”。研究时间跨度为五年,从2020年10月1日到2025年9月30日。
社会合作课程是根据私营组织的资金而设立和运行的课程,这些私营组织致力于与东京大学针对共同关注公共问题展开研究合作。
在东京大学研究生院药学院,将开设“发现蛋白质降解药物”课程。国家健康科学研究所分子靶标和基因治疗产品部前主任mikihiko naito博士被任命为该项目的项目教授,并将领导包括sniper在内的蛋白质降解技术的研究。这项研究将结合研究生院进行的世界上最先进的泛素蛋白酶体研究与卫材培养的药物发现知识,以便开发针对药物靶向蛋白质的新蛋白质降解技术,并促进基于该技术的药物发现研究。此外,通过这项研究,该课程还将教育和培养该研究领域的下一代领导人。
靶向蛋白质降解是一系列技术,其中精确设计的化合物迫使靶蛋白质接近e3泛素连接酶,并应用泛素蛋白酶体系统来诱导靶蛋白质降解。该技术不仅为常规靶标(如特异性酶和受体),还为迄今为止难以发现药物的疾病相关蛋白质提供一种药物制备方法。通过这项技术开发和药物发现,东京大学和卫材的目的是为先前治疗选择有限的患者提供新治疗选择。
[编辑注释]
sniper(特异性和非遗传性iap-依赖性蛋白质擦除器)是一种利用泛素蛋白酶体系统降解靶蛋白质的化合物。该化合物是一种“混合化合物”,并由一个与靶蛋白质结合的部分和一个通过合适的连接键与e3泛素连接酶(iap)结合的部分组成。设计该化合物需要先进的药物化学和最新的结构生物学。当sniper化合物使靶蛋白质和e3泛素连接酶(iap)接近时(下图中的步骤1),泛素作为蛋白质降解标签从e2泛素结合酶转移到靶蛋白质(2),用于蛋白酶体识别蛋白质及随后的降解(3)。
常规小分子抑制剂与靶酶的活性部分结合,并通过抑制其活性来表达药理活性。另一方面,由于sniper通过上述靶蛋白降解表现出药理特性,不仅预计其表现出与小分子抑制剂不同的药理活性;还预测其靶向不具酶活性的蛋白质。类似的技术包括protac(蛋白水解靶向嵌合体)和degronimid。
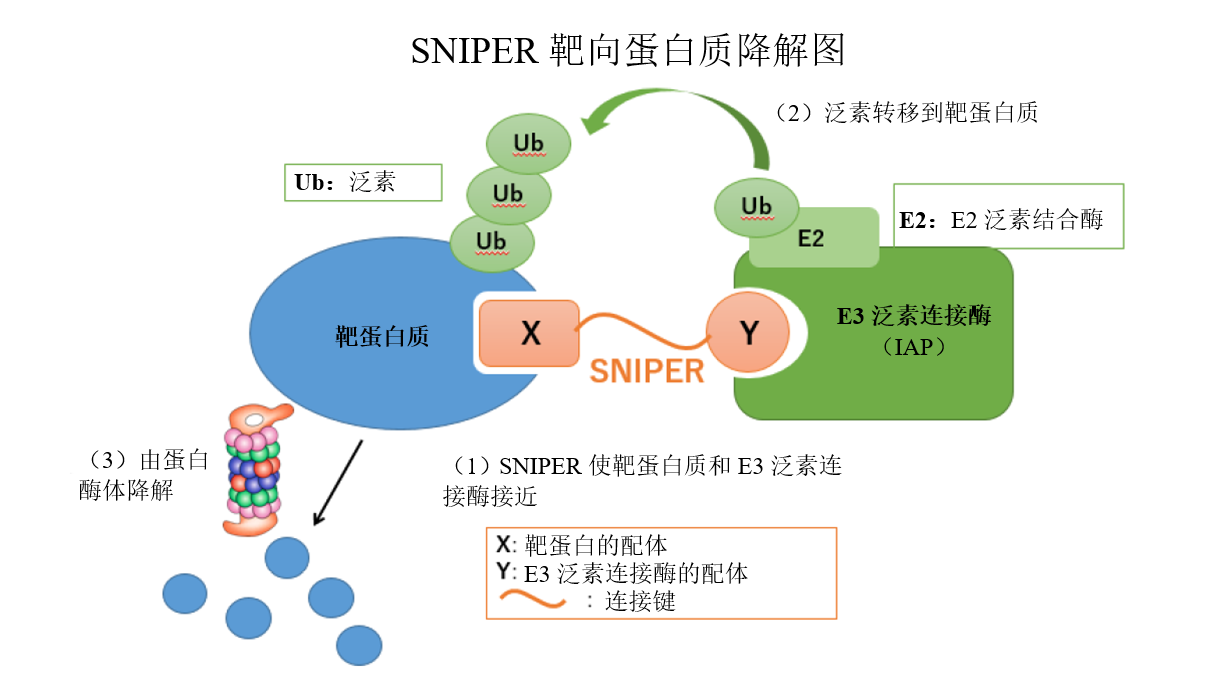
关于泛素蛋白酶体系统
泛素蛋白酶体系统是控制不需要的蛋白质降解的自然发生机制之一,并控制与生命保护相关的关键运动,包括细胞周期、转录调控和信号传递。当泛素作为蛋白质降解标签通过试剂(如e3泛素连接酶)连接到不需要的蛋白质上时,其标记该蛋白质,用于蛋白酶体识别及随后的降解。近年来,泛素蛋白酶体系统与人类主要疾病(包括癌症和神经退行性变)之间的关系日益明显。
在amed公开招募的项目“开发用于新型冠状病毒感染(covid-19)的治疗药物”中被采纳
eisai co., ltd.(总部:东京,首席执行官:内藤晴夫,以下简称“eisai”)近日宣布,公司已与日本的四个研究组织(kan research institute、日本国立国际医疗研究中心、长崎大学和横滨市立大学)就“开发预防新型冠状病毒传染性疾病(covid-19)恶化的治疗药物”(授权号:20fk0108255)签订了一份联合研究协议,这是以eisai为代表研究组织的研究项目之一。该联合研究项目在日本医疗研究开发机构(amed)2020财政年度促进新发和重新出现的传染性疾病创新治疗研究和开发业务“开发用于新型冠状病毒传染性疾病(covid-19)的治疗药物”项目的第二次公募中被采纳。
在因sars-cov-2感染而出现covid-19的患者中,已报告出现例如急性呼吸窘迫综合征(ards)和随后出现的多器官衰竭等重症病例。研究人员假定血管病的形成和加重以及细胞因子风暴*参与了疾病恶化过程,但目前尚未完全了解基于sars-cov-2感染的恶化机制。
在该项合作研究中,将构建sars-cov-2感染的非临床动物模型。此外,将对eisai发现的tlr(toll样受体)4拮抗剂eritoran和kan research institute(eisai的研究子公司)发现的抗fkn(fractalkine)抗体e6011进行评估。同时,该项目将促进使用来源于sars-cov-2感染患者的临床样本进行生物标志物研究。该项合作研究旨在阐明基于sars-cov-2感染的covid-19恶化机制,并开发预防covid-19恶化的药物。
在抗击covid-19蔓延的过程中,eisai将基于关心人类健康(hhc)理念,继续在各国进行治疗药物开发、稳定药物供应并支持相关活动。
*细胞因子风暴:一种免疫失控状态,在这种状态下,发挥激活免疫应答作用的细胞因子的产生变得无法控制,进而释放出大量细胞因子。
[编者注]
1.tlr4和eritoran(e5564)简介
tlr(toll样受体)是先天免疫系统的受体,并可识别病原体的特定分子结构。研究认为,由tlr启动激活的先天免疫系统在消除病原体、引起炎症反应或抗病毒反应中起关键作用。tlr4是tlr(构成各种受体家族)家族中的一员,可被细菌释放的脂多糖等内毒素激活。eritoran是eisai公司内部通过天然产物有机合成技术发现并开发的tlr4拮抗剂。它是类脂a的结构类似物,类脂a是内毒素的活性药效成分。在14项临床研究(包括一项在重度脓毒症中进行的大型iii期随机试验)中,既往观察到该抗结剂具有耐受良好的安全性特征。在小鼠流感病毒感染模型中,eritoran具有抑制细胞因子产生和改善全身状况的作用1。预期可通过抑制tlr4的激活来抑制covid-192,3引起的炎症和恶化;tlr4是引起细胞因子风暴的各种细胞因子基因表达信号传递的最上游。
在中度covid-19住院患者的国际试验remap-covid中,已选择eritoran作为候选治疗药物。
2.fkn和e6011简介
fkn(fractalkine)是一种具有细胞迁移调节和细胞粘附双重功能的趋化因子,在炎症过程中诱导血管内皮细胞。fkn受体(cx3cr1)主要在单核细胞、巨噬细胞和杀伤淋巴细胞中选择性表达,在将相关细胞高效募集到炎症部位方面起关键作用。有意见认为,fkn-cx3cr1系统与多种慢性炎症性疾病有关,包括炎症性肠病、类风湿性关节炎、肝脏疾病、中枢神经系统疾病、动脉硬化、皮肤病等。e6011是由kan research institute, inc.(eisai的研究子公司)开发的世界首个人源化抗fkn单克隆抗体,与现有细胞因子治疗不同,e6011具有新作用机制,即通过中和fractalkine(fkn)活性来抑制细胞侵袭。目前,ea pharma co., ltd.(eisai的胃肠疾病子公司)正在克罗恩病患者中开展一项ii期临床试验。e6011可抑制cd16 单核细胞(cx3cr1高表达的细胞群,对局部炎症反应起重要作用)与血管内皮细胞紧密结合4。因此,预期e6011可抑制covid-19中的血管病变的发生和恶化5。
1 ka shirey et al., nature.2013 may 23; 497(7450):498-502
2 p mehta et al., the lancet 2020; 395: 1033-1034
3 c huang et al., the lancet 2020; 395: 497-506
4 y kuboi et al., int immunol. 2019 apr 26;31(5):357
5 h li et al., the lancet 2020; 395: 1517-1520
卫材株式会社(总部:东京,ceo:内藤晴夫,下称“卫材”)和生化学工业株式会社(总部:东京,总裁:水谷建,“生化学”)近日宣布,两家公司已就生化学首创的骨关节炎治疗剂si-613(双氯芬酸结合透明质酸钠)在韩国的联合上市签订协议。这是自卫材和生化学于2020年4月1日就si-613在中国的共同开发和联合上市签署协议以来,缔结联合上市协议的第二个国家。
根据该协议,eisai korea inc.(卫材子公司)将获得si-613在韩国的独家上市权,并申请其制造和上市许可。获批后,生化学将向卫材供应产品,而卫材将负责分销。卫材将向生化学支付预付款,并达到销售里程碑。
骨关节炎是一种由老化和其他因素导致的关节软骨损伤引起的疾病,引发炎症和疼痛,从而导致生活质量(qol)下降。膝骨关节炎是其疾病中最常见的病例之一,韩国膝骨关节炎患者的人数估计约为320万。*1预计随着人口老龄化,该数字将继续增加。
si-613是由生化学利用其专有药物结合技术将透明质酸和双氯芬酸(一种抗炎药物)化学结合而成的双氯芬酸结合透明质酸钠。这种物质具有双氯芬酸的止痛和抗炎作用,除透明质酸钠的关节功能改善作用外,还旨在通过释药系统*3起到缓释作用*2。因此,预计si-613可快速且持续减轻与骨关节炎相关的疼痛和炎症。
根据该协议,卫材旨在通过利用其韩国业务获得的知识和网络,满足膝骨关节炎患者未得到满足的医疗需求。生化学将通过利用卫材的韩国业务基地,寻求在韩国实现si-613价值的最大化。
两家公司将通过si-613的商业化,在韩国提供治疗膝骨关节炎的新治疗方案,以促进改善患者的qol。
<编辑注释>
1. 关于si-613
si-613是由生化学利用其独有的药物结合技术,将透明质酸和双氯芬酸(一种抗炎药物)化学结合而成的双氯芬酸结合透明质酸钠。这种物质具有双氯芬酸的止痛和抗炎作用,除透明质酸钠的关节功能改善作用外,还旨在通过释药系统*3起到缓释作用*2。预计si-613可快速且持续减轻与骨关节炎(如膝关节)相关的疼痛和炎症。另外,由于通过注射将其直接给药至关节腔中,认为双氯芬酸全身暴露量和引起全身药物不良反应的风险低。
2020年1月6日,生化学提交一份新药申请(下称“nda”),申请批准在日本制造和上市si-613以治疗骨关节炎(膝关节、髋关节和踝关节)。卫材和生化学于2020年4月1日就si-613在中国的共同开发和上市联盟签署协议。
2. 关于卫材株式会社
卫材株式会社将企业使命定义为“我们将患者及家属的利益放在首位,为提升其福祉做出贡献”,即为关心人类健康(hhc)理念。遍布全球的研发机构、生产基地和营销附属公司总共拥有近10,000名员工。我们努力实践hhc理念,通过提供创新产品满足待满足的医疗需求,特别是在肿瘤学和神经学领域。作为一家全球制药公司,我们的使命是通过与主要利益相关方合作,改善发展中国家和新兴国家的药品获取渠道,从而使世界各地的患者受益。
欲了解更多有关卫材株式会社的信息,请访问https://www.eisai.com
3. 关于生化学工业株式会社
生化学工业株式会社是一家研发型制药公司,专注于糖科学作为专业领域。自1947年创立以来,生化学一直专注于糖科学的可能性,并在骨科疾患和眼科疾病领域开发原创且有益的药物产品和医疗器械。在独特业务模式(即专注于研发和制造,而无内部药品销售部门)下,生化学通过与在特定国家和产品领域具有优势的公司合作,在全球范围内将产品上市,为全世界人民的健康和充实的生活做出贡献。
欲了解更多有关生化学工业株式会社的信息,请访问https://www.seikagaku.co.jp/en/
参考文件:
*1: for the estimated data regarding the number of patients with knee osteoarthritis
· vital data – korea statistical information service, url:http://kosis.kr/index/index.do
· changes in the number of patients – healthcare bigdata hub, url:https://opendata.hira.or.kr/home.do
*2: sustained release is a gradual release of the active pharmaceutical ingredients of a drug to achieve a sustained therapeutic effect.
*3: drug delivery system (dds) is a technology for the controlled release, targeting, and absorption improvement of drugs.
–在临床试验中,证实52周内非戈替尼的疗效持久和安全性一致-
吉利德科学公司(纳斯达克:gild)和卫材株式会社(日本东京)近日宣布,日本厚生劳动省(mhlw)已准予吉利德 k.k.(日本东京)对jyseleca®(非戈替尼200mg和100mg片剂)的监管批准。jyseleca®是一种每日一次的口服jak1优先抑制剂,用于治疗对常规疗法反应不足的患者的类风湿性关节炎(ra),包括预防结构性关节损伤。
吉利德日本将拥有jyseleca在日本的销售授权,并将负责日本jyseleca产品供应,而卫材将负责日本jyseleca(用于治疗ra)产品分销。两家公司将共同商售这种药物,以供日本医生和患者使用。
“尽管ra治疗取得进展,但现有疗法并不能使许多患者达到临床指南中推荐的治疗目标。继续需要有效且耐受性良好的新治疗方案,” 庆应义塾大学医学院内科教授兼风湿病学专家tsutomu takeuchi(医学博士)称。“jyseleca是一种新jak抑制剂,在临床试验中,在广泛的患者群体(包括对生物制剂反应不足的患者)中,显示出临床改善、疾病活动性低和临床缓解。”
“ra导致许多患者虚弱、疲劳和疼痛,这可能会严重干扰其日常生活,”职业与环境卫生大学第一内科教授yoshiya tanaka(医学博士)称。“重要的是存在新治疗方案,可有效控制患者的症状,并为其带来新希望。”
基于全球finch 3期和darward 2期计划的稳健临床试验结果,获得日本的批准。finch和darwin计划评估jyseleca对ra患者群体(包括初治患者和经证明对包括生物dmard在内的护理标准治疗反应不足的患者)中3,500多名患者的疗效。每日接受一次jyseleca的患者的临床体征和症状有所改善、疾病活动减少、以及关节结构损伤进展较少。
在整个finch试验中,证明jyseleca的安全特性一致,并且关注的不良事件(包括严重感染、带状疱疹、静脉血栓栓塞和主要心血管事件)的频率与对照组的相当。
在整个finch和darward试验中,最常见的不良反应是恶心、上呼吸道感染、尿路感染和头晕。带状疱疹和肺炎发生率分别为0.2%和0.3%。jyseleca 200mg组和jyseleca 100mg组每年每100人严重感染的暴露调整发生率(95% ci)分别为1.7%(1.3,2.1)和2.5%(1.9,3.3)。开jyseleca处方时,建议医生监测患者新严重感染的发展或现有严重感染的恶化,包括肺炎、肺结核、败血症和其他病毒感染。
“这项监管批准承认jyseleca将有望为尚未成功接受既往疗法治疗的ra患者提供的益处,并代表这一挑战性疾病治疗方面的重要进展,” 吉利德科学,k.k.的总裁兼代表董事luc hermans(医学博士)称。
“现在,jyseleca已获得日本的批准,我们期待利用自己在日本ra领域临床开发和商售的丰富经验,尽快将这一新治疗方案带给日本患者,并为提高患者生活质量做出贡献,”卫材高级副总裁日本卫材总裁hidenori yabune称。
吉利德正在与加拉帕戈斯群岛公司(比利时梅赫伦(纳斯达克和泛欧:glpg))合作开发jyseleca。这两家公司正在进行全球研究,研究jyseleca在各种疾病中的潜在作用,包括先前报告的有关溃疡性结肠炎的3期selection试验。
关于finch计划
finch 3期计划研究每日一次非戈替尼100mg和200mg对ra患者群体(从早期到具有生物学经验的患者)的疗效和安全性。finch 1是一项与mtx联合进行的为期52周的随机、安慰剂和阿达木单抗对照试验,招募1,759名对mtx治疗反应不足的成年中重度活动性ra患者。finch 1的主要终点是第12周时的acr20。该试验包括第24周和第52周时的放射学评估。finch 2是一项为期24周的全球性随机、双盲、安慰剂对照3期研究,以常规合成疾病缓解性抗风湿药(csdmard)为背景,对449名对生物dmard(bdmard)反应不足的成年中重度活动性ra患者评估非戈替尼。finch 2的主要终点是第12周时的acr20。finch 3是一项为期52周的随机试验,对1,252名mtx-初治患者评估单独给药非戈替尼200mg和非戈替尼100mg或200mg联合mtx与单独给药mtx的疗效。finch 3的主要终点是第24周时的acr20。该试验包括第24周和第52周时的放射学评估。
关于吉利德科学
吉利德科学公司是一家以研究为基础的生物制药公司,在未满足医疗需求的领域发现、开发和商售创新药物。该公司致力于转变和简化对全球绝症患者的关心。吉利德在全球超过35个国家开展业务,总部位于加利福尼亚州福斯特市。
欲了解更多关于吉利德科学的信息,请访问公司网站:www.gilead.com。
欲了解更多关于吉利德科学k.k.的信息,请访问公司网站:https://www.gilead.co.jp/。
关于卫材株式会社
卫材株式会社是一家总部位于日本的领先全球制药公司。企业使命是“我们将患者及家属的利益放在首位,为提升其福祉做出贡献”,称之为关心人类健康(hhc)理念。我们拥有将近10,000名员工,凭借研发机构、生产基地和销售附属公司的全球网络,我们致力于通过针对医疗需求未得到充分满足的疾病提供创新产品,实现我们的关心人类健康理念,尤其侧重于我们的神经学和肿瘤学战略领域。作为一家全球性制药公司,我们的使命造福世界各地的患者,我们通过投资和参与基于伙伴关系的倡议,改善发展中国家和新兴国家获得药品的机会。
欲了解更多关于卫材株式会社的信息,请访问https://www.eisai.com
吉利德前瞻性声明
本新闻稿包括1995年《私人证券诉讼改革法案》意义内的前瞻性声明-受到风险、不确定性和其他因素的影响,包括在日本无法成功商售jyseleca用于治疗类风湿性关节炎的风险。涉及jyseleca的进行中和额外临床试验也可能会产生不利结果,并且存在其他监管机构可能不批准jyseleca用于治疗类风湿性关节炎和其他适应症的风险,并且任何上市许可(如果准予)均可对其使用有重大限制。此外,吉利德可能会做出战略决策,中止开发和商售jyseleca,因此,可能永远无法成功商售jyseleca。这些风险、不确定性和其他因素可能导致实际结果与前瞻性声明中提及的结果明显不同。告诫读者不要依赖这些前瞻性声明。在吉利德向美国证券交易委员会提交的截至2020年6月30日的季度表10-q上的季度报告中,详细描述这些和其他风险。所有前瞻性声明均基于吉利德目前掌握的信息,并且吉利德不负责更新任何此类前瞻性声明。
jyseleca®、吉利德和吉利德标志是吉利德科学公司或其相关公司的商标。
卫材株式会社(总部:东京,ceo:内藤晴夫,“卫材”)宣布,其英国子公司提交的关于其独家研发的抗癫痫药物(aed)fycompa®(通用名:吡仑帕奈)儿科适应症的许可证扩展申请,已获得欧洲药品管理局(ema)人用药品委员会(chmp)的肯定意见。chmp的肯定意见是,通过将经批准的年龄范围从12岁及以上扩展到4岁及以上,以及将原发性全面性强直-阵挛发作(pgtcs)的年龄范围从12岁及以上扩展到7岁及以上,扩展fycompa的使用范围,作为部分性癫痫发作(pos)(伴或不伴继发性全面性发作)的辅助治疗药物。
该申请(于2019年2月递交ema)基于全球范围内进行的iii期(研究311)和ii期(研究232)临床研究的结果。这些研究评估fycompa作为pos或pgtcs儿科患者的辅助治疗药物。研究311评估fycompa作为辅助治疗药物给药时用于4岁至12岁以下pos或pgtcs控制不当的儿科患者的安全性、耐受性和暴露-疗效关系。研究232评估fycompa作为辅助治疗药物用于儿科癫痫患者(2岁至12岁以下)的药代动力学、疗效和长期安全性。
fycompa是由卫材独家研发的一种新型抗癫痫药物。癫痫发作是由神经递质谷氨酸介导的。该药是一种高选择性、非竞争性的 ampa 型受体拮抗剂,可通过靶向突触后细胞膜上的ampa受体处的谷氨酸活性,减少与癫痫发作相关的神经元的过度兴奋。在日本,fycompa适用于4岁及以上癫痫部分性发作(伴或不伴继发性全面性癫痫发作)患者的单药治疗和辅助治疗。及作为12岁及以上癫痫患者的原发性全面强直阵挛发作性癫痫的辅助治疗。此外,在美国,fycompa也适用于4岁及以上癫痫部分性发作(伴或不伴继发性全面性癫痫发作)患者的单药治疗和辅助治疗。及作为12岁及以上癫痫患者的原发性全面强直阵挛发作性癫痫的辅助治疗。
卫材认为包括癫痫在内的神经学是一个重点治疗领域。随着fycompa在欧洲的上市,卫材致力于让世界各地更多癫痫患者实现“零发作”的目标。卫材致力于满足癫痫患者及其家人的各种需求,并为其提供更多福利。
媒体咨询:
卫材株式会社
公共关系部
81-(0)3-3817-5120
[编者注]
fycompa是由卫材独家研发的一种新型抗癫痫药物。癫痫发作是由神经递质谷氨酸介导的,该药物是一种高选择性、非竞争性的ampa受体拮抗剂,通过靶向突触后膜上ampa受体处的谷氨酸盐活性,减少与癫痫发作相关的神经元的过度兴奋。fycompa制剂现已上市销售,每日一次,睡前口服。日本已批准片剂和细颗粒制剂。该药的口服混悬剂和片剂已在美国和欧洲获得批准。
目前,fycompa已在70多个国家和地区,包括日本、美国、中国、以及欧洲和亚洲的其他国家,作为12岁及以上癫痫患者的部分性癫痫发作(伴或不伴继发性全面性癫痫发作)的辅助治疗。此外,fycompa已被美国、日本、欧洲和亚洲等65多个国家批准作为12岁及以上癫痫患者的原发性全面强直阵挛发作性癫痫的辅助治疗。在日本和美国,fycompa获批用于单药治疗和辅助治疗4岁及以上癫痫患者的部分性癫痫发作(伴或不伴继发性全面性癫痫发作)。迄今为止,fycompa已被广泛应用于治疗全球超过300,000名患者(所有适应症合计)。卫材正在全球范围内进行一项iii期临床研究(研究338),意在将该药用于治疗与林-戈(lennox-gastaut)综合征相关的癫痫发作。此外,卫材注射制剂的开发也在进行中。
研究311是一项全球性(美国、欧洲、日本、韩国)开放性iii期临床研究,评估fycompa口服混悬液作为辅助治疗药物给药时用于180名4岁至12岁以下部分性癫痫发作或原发性全身强直阵挛发作控制不当的儿童癫痫患者的安全性、耐受性和暴露疗效关系。这项研究包含一个治疗期(包括长达11周的滴定期和长达12周的维持期)以及一个延长期。在这项研究中,每日睡前口服一次2~16mg fycompa。主要终点是安全性和耐受性。疗效与对12岁及以上患者观察到的相似。在这项研究中观察到的最常见的不良事件(发生率为10%或更高)是嗜睡、鼻咽炎、发热、呕吐、头晕、流行性感冒、以及易怒,这与迄今为止fycompa的安全特性一致。
研究232是一项全球性(美国、欧洲)多中心、开放标签临床研究,具有一个延长期,旨在评估63名儿科癫痫患者(2岁至12岁以下)。该研究评估与其他aed同时服用的fycompa口服混悬液的药代动力学、安全性、耐受性和疗效。每日给药一次fycompa的滴定剂量范围为0.015mg/kg~0.18mg/kg,并且经过11周的治疗和一个延长期(41周)后,确认长期安全性。在研究232中观察到的不良事件(fycompa组≥10%)为发热、疲劳、呕吐、易怒、嗜睡、头晕、上呼吸道感染。
癫痫按癫痫发作类型大致进行分类,部分发作性癫痫发作约占癫痫病例的60%,全身性癫痫发作约占40%。在部分发作性癫痫发作中,异常的电干扰发生在脑的有限区域,并且可能随后扩散到整个脑,从而成为全身性癫痫发作(称为继发性全身性癫痫发作)。在全身性癫痫发作中,异常的电干扰发生在整个脑,随后可能出现意识丧失或全身出现身体症状。
癫痫在日本影响大约100万人、在美国影响340万人、在欧洲影响600万人、在中国影响900万人、在全世界影响大约6000万人。
由于约30%的癫痫患者无法使用现有的aeds3控制癫痫发作,这是一种未充分满足医疗需求的疾病。虽然任何年龄的人都会发作,但18岁及以下的人和老年人最常发作。由于儿科癫痫的病因和临床症状不统一,并且预后范围可能从非常积极的病例到顽固的病例,需要特别考虑对每名患者的治疗。
1 a. fogarasi et al. 开放性研究-旨在研究辅助治疗药物吡仑帕奈用于儿童(4岁至<12岁)局灶性癫痫发作或全身强直阵挛性癫痫发作的安全性和有效性。2020年1月;第61(1)期:第125-137页。
2 j. ben renfroe et al. 辅助治疗药物吡仑帕奈口服混悬剂用于≥2岁至<12岁儿科癫痫患者:药代动力学、安全性、耐受性和疗效。j child neurol2019年4月;第34(5)期:第284-294页
3 “癫痫和癫痫发作:研究期望。什么是癫痫?”国立神经病学与中风研究所,2016年5月24日访问,http://www.ninds.nih.gov/disorders/epilepsy/detail_epilepsy.htm#230253109
新结果包括ii期leap-004试验的结果,该试验表明联合疗法用于既往接受抗pd-1/pd-l1治疗后出现疾病进展的不可切除或晚期黑色素瘤患者的orr为21.4%
卫材株式会社(总部:东京,首席执行官:内藤晴夫,以下简称“卫材”)和merck & co., inc.(kenilworth, n.j.,美国;美国和加拿大以外国家和地区称为msd)宣布了leap(仑伐替尼联合帕博利珠单抗)系列临床研究计划中评价lenvima(卫材研发的口服多靶点酪氨酸激酶受体抑制剂)联合keytruda(默沙东公司的抗pd-1治疗药物)的两项试验的最新研究数据。在ii期leap-004试验中,在既往接受抗pd-1/pd-l1治疗后出现疾病进展的不可切除或晚期黑色素瘤患者中,lenvima联合keytruda的客观缓解率(orr)为21.4%(95%置信区间[ci]:13.9-30.5)。在ii期leap-005试验中,用于既往经治的三阴性乳腺癌(tnbc)、卵巢癌、胃癌、结直肠癌(非微卫星高度不稳定性[非msi-h]/错配修复基因正常[pmmr])、多形性胶质母细胞瘤(gbm)和胆管癌(btc)患者中,lenvima联合keytruda的orr范围为9.7-32.3%(95% ci:2.0-51.4)。leap-004(摘要号lba44)和leap-005(摘要号lba41)的结果被2020年欧洲肿瘤内科学会(esmo)年会接收为延迟公布摘要,并在会上进行展示和汇报。
默沙东研究实验室临床研究副总裁scot ebbinghaus博士称:“这些来自leap临床项目的新数据表明,keytruda联合lenvima在几种侵袭性癌症类型中表现出鼓舞人心的抗肿瘤活性,让我们更深入的了解了该联合用药的潜力,这将帮助更多患有这些癌症的患者”。同时scot ebbinghaus博士也称:“这是我们首次展示两项leap试验的临床数据,这些数据反映了我们目前在探索这种联合用药对需要新治疗选择的患者的潜力,特别是对那些既往接受抗pd-1或pd-l1治疗后出现疾病进展的晚期黑色素瘤患者的潜力方面所取得的重大进展。”
卫材副总裁、肿瘤事业部首席医学创新官(chief medicine creation)兼首席发现官(chief discovery officer)takashi owa博士称:“迄今为止,在13种不同癌症中获得的越来越多的研究成果使我们受到鼓舞,这些研究成果支持lenvima与keytruda联合用药的潜力,目前,我们正在19项临床试验中对这种联合用药进行评价”。同时takashi owa博士也称:“这些数据不仅有助于我们加深对治疗方案的了解,而且更加坚定了我们的决心,努力解决这些患者未满足的医疗需求。”
仑伐替尼(len)联合帕博利珠单抗(pembro)治疗接受pd-1或pd-l1抑制剂治疗后出现疾病进展的晚期黑色素瘤(mel):leap-004(摘要号lba44)的初步结果
leap-004(clinicaltrials.gov,nct03776136)是一项在12周内接受抗pd-1/pd-l1治疗后出现疾病进展的不可切除或晚期黑色素瘤患者中评价lenvima联合keytruda的ii期、单臂、开放性试验。患者口服lenvima 20 mg,每日一次,同时静脉输注keytruda 200 mg,每3周一次,持续35个周期(约2年),直至出现不可耐受的毒性或疾病进展。主要终点为orr,由设盲独立中心审查(bicr)根据实体瘤疗效评价标准(recist)v1.1进行评估。次要终点包括无进展生存期(pfs)和缓解持续时间(dor)(由bicr根据recist v1.1评估)、总生存期(os)以及安全性。
截至数据截止日期(2020年6月10日),共有103例患者入组并接受治疗。中位随访持续时间为12个月(范围:8.7-15.6),lenvima联合keytruda的总体orr(由bicr评估)为21.4%(n=22)(95% ci:13.9-30.5),完全缓解率为1.9%(n=2),部分缓解率为19.4%(n=20)。在总体研究人群中,中位dor为6.3个月(范围:2.1 至11.1 ),72.6%(95% ci:46.2-87.6)的患者缓解持续至少6个月。中位pfs为4.2个月(95% ci:3.5-6.3),73.8%的患者出现疾病进展或死亡,9个月pfs率为26.2%(95% ci:17.4-35.9)。中位os为13.9个月(95% ci:10.8-未达到[nr]),44.7%的患者死亡,9个月os率为65.4%(95% ci:55.2-73.8)。
探索性分析表明,具体来说,在接受抗pd-1/l1治疗 抗ctla-4治疗后出现疾病进展的29例患者中,orr(由bicr评估)为31%(95% ci:15.3-50.8),完全缓解率为3.4%(n=1),部分缓解率为27.6%(n=8)。在这些患者中,疾病控制率(dcr,由bicr评估)为62.1%(95% ci:42.3-79.3)。在总体研究人群中,dcr(由bicr评估)为65%(95% ci:55.0-74.2)。
治疗相关不良事件(trae)导致7.8%的患者终止lenvima和/或keytruda治疗。44.7%的患者发生3-5级trae(3级:39.8%;4级:3.9%;5级:1.0%),18.4%的患者发生严重trae。总体研究人群发生的最常见(≥30%)的任何级别的trae为高血压(56.3%)、腹泻(35.9%)、恶心(34.0%)、甲状腺功能减退症(33.0%)和食欲减退(31.1%)。
leap-005:仑伐替尼联合帕博利珠单抗用于治疗既往经治晚期实体瘤患者的ii期研究(摘要号lba41)
leap-005(clinicaltrials.gov,nct03797326)是一项在选定既往经治晚期实体瘤患者中评价lenvima联合keytruda的ii期、单臂、开放性试验。研究队列为tnbc、卵巢癌、胃癌、结直肠癌(非msi-h/pmmr)、gbm和btc患者。患者口服lenvima 20 mg,每日一次,同时静脉输注keytruda 200 mg,每3周一次,持续35个周期(约2年),直至出现不可耐受的毒性或疾病进展。主要终点为orr(由bicr根据recist v1.1或神经肿瘤学疗效评估[rano]标准[仅适用于gbm]评估)和安全性。次要终点包括dcr(由bicr根据recist v1.1或rano[仅适用于gbm]评估)、dor(由bicr根据recist v1.1或rano[仅适用于gbm]评估)、pfs(由bicr根据recist v1.1或rano[仅适用于gbm]评估)以及os。
截至数据截止日期(2020年4月10日),共有187例患者入组并接受治疗。中位随访持续时间为8.6个月(范围:1.9-13.1)时,6种不同肿瘤类型证实的orr以及有效性和安全性结果:
总体研究人群发生的最常见(≥20%)的任何级别的trae为高血压(39.0%)、疲乏(29.4%)、腹泻(26.7%)、食欲减退(25.1%)、甲状腺功能减退症(27.8%)和恶心(21.9%)。基于这些初步结果,该试验将扩展至每个队列入组约100例患者。
lenvima®(仑伐替尼)胶囊简介
lenvima是由卫材发现并开发的一种多靶点酪氨酸激酶受体抑制剂,可抑制血管内皮生长因子(vegf)受体vegfr1(flt1)、vegfr2(kdr)和vegfr3(flt4)的激酶活性。除正常细胞功能外,lenvima还可抑制与致病性血管生成、肿瘤生长和癌症进展相关的其他激酶,包括成纤维细胞生长因子(fgf)受体fgfr1-4、血小板衍生生长因子受体α(pdgfrα)、kit和ret。在同系小鼠肿瘤模型中,与任一药物单独给药相比,仑伐替尼联合抗pd-1抗体可减少肿瘤相关巨噬细胞,增加活化的细胞毒性t细胞,并表现出更强的抗肿瘤活性。目前,lenvima已在超过65个国家(包括日本、美国、欧洲和亚洲的部分国家)获批单药用于甲状腺癌的治疗,并在超过65个国家(包括日本、美国、欧洲部分国家、中国和亚洲部分其他国家)获批单药用于不可切除肝细胞癌的治疗。此外,lenvima也在超过55个国家(包括美国、欧洲部分国家[以商品名kisplyx®上市,用于治疗肾细胞癌]和亚洲部分国家)获批与依维莫司联合用于既往接受过抗血管生成治疗的晚期肾细胞癌患者。同时,lenvima也在包括美国、澳大利亚和加拿大在内的国家获批用于与keytruda联合用药治疗在既往接受全身治疗后出现疾病进展,不适合根治性手术或放疗的非微卫星高度不稳定(msi-h)或错配修复缺陷(dmmr)型晚期子宫内膜癌患者。该适应症的持续批准取决于各项确证性试验中临床获益的验证和描述。
keytruda®(帕博利珠单抗)注射液简介
keytruda是一种抗pd-1治疗药物,通过提高人体免疫系统帮助发现和抗击肿瘤细胞的能力发挥作用。keytruda是一种人源化单克隆抗体,可阻断pd-1与其配体pd-l1和pd-l2之间的相互作用,从而激活可能影响肿瘤细胞和健康细胞的t淋巴细胞。
默沙东目前正在进行行业内规模最大的免疫肿瘤学临床研究项目。目前有1200多项在多种癌症和治疗环境中研究keytruda的试验。keytruda临床项目旨在了解keytruda在癌症中的作用以及可能预测患者从keytruda治疗中获益可能性的因素,包括探索几种不同的生物标志物。
卫材和默沙东战略合作简介
2018年3月,卫材和默沙东通过其子公司展开了一项关于lenvima全球联合开发和联合商业化的战略合作。根据协议,两家公司将联合开发、生产和商业推广lenvima,既可单药治疗也可与默沙东抗pd-1治疗药物keytruda联合用药。
除了在几种不同肿瘤类型中正在进行的评价keytruda与lenvima联合用药的临床研究外,公司还通过leap(仑伐替尼联合帕博利珠单抗)临床项目共同发了起新的临床研究,正在13种肿瘤类型(子宫内膜癌、肝细胞癌、黑色素瘤、非小细胞肺癌、肾细胞癌、头颈部鳞状细胞癌、尿路上皮癌、胆道癌、结直肠癌、胃癌、胶质母细胞瘤、卵巢癌和三阴性乳腺癌)中通过19项临床试验对该联合用药进行评价。
卫材专注于癌症
卫材专注于抗癌药物开发并将所有知识和经验应用于肿瘤微环境,包括halaven®(甲磺酸艾立布林)和lenvima®、驱动基因突变以及利用rna剪切平台的异常剪接等领域(ricchi),这些领域还有很多患者的需求未得到真正满足而且卫材希望成为肿瘤学领域的领导者。卫材将从这些ricchi中发现具有新的靶点和新的作用机制的创新药并致力于癌症治疗。
卫材简介
卫材是一家领先的全球研发制药公司,总部位于日本,在全球约有10000名员工。卫材将企业使命定义为“将患者及家属的利益放在首位,为提升其福祉做出贡献”,公司将这种使命称为关心人类健康(hhc)理念。卫材致力于在肿瘤学和神经病学等医疗需求高度未得到满足的治疗领域提供创新产品,努力实现公司的hhc理念。本着hhc的精神,卫材会进一步履行这一承诺,运用公司拥有的科学专业知识、临床能力和对患者的了解,发现并开发创新的pg电子app的解决方案,帮助解决社会最棘手的未得到满足的需求,包括被忽视的热带病和可持续发展目标。
有关卫材的更多信息,请访问 www.eisai.com(适用于全球)、us.eisai.com(适用于美国)或(适用于欧洲、中东、非洲),并通过twitter(&
2020年9月20日,由卫材(中国)药业有限公司(以下简称“卫材中国药业”)主办的“2020 ads峰会”(ads全称advance dementia science)成功举办。本次大会以“run into the next decade(奔向下一个十年)”为主题,分为一个主会场(线上)与北京、上海、广州、成都四地分会场(线下),来自中、日、韩三国知名专家和线上、线下近千位嘉宾共话痴呆与认知障碍前沿进展。ads峰会是卫材中国药业倾力打造的又一医生信息交流平台,自2018年首届ads峰会举办以来,今年已是第三届。
峰会伊始,在主会场上,首先由卫材全球高级副总裁、卫材中国总裁冯艳辉女士致辞。冯艳辉表示,热烈欢迎各位专家和参会嘉宾出席本次“2020 ads峰会”,并衷心感谢各位对此次峰会的大力支持。卫材中国药业将致力于打造认知障碍学术交流平台,持续在该领域加大新药研究与开发。同时,以 hhc(human health care,“关心人类健康”)企业理念,与社会各界共同在疾病认知、预防与控制、患者关爱等多方面做出贡献。

冯艳辉在主会场(线上)致辞
在此次峰会上,由来自首都医科大学宣武医院的贾建平教授受邀担任大会主席。贾建平教授表示,感谢卫材中国搭建这样一个学术交流平台,使中、日、韩专家和参会嘉宾一同进行学术前沿进展与临床实践方面的分享,并预祝峰会圆满成功。
之后,由来自韩国建国大学附属医院的moon yeon-sil教授与来自日本昭和大学附属医院的小野贤二郎教授分别带来“最‘in’的ad生物标记物和应用”和“ad新药乘风破浪,谁将第一个到达彼岸”的主题分享,为线上线下观众带来专业且深度的领域内见解。
在北京、上海、广州、成都四地的线下分会场上,设置了“2020aaic热点精粹”、“ad的全程管理”等痴呆与认知障碍领域内的热点与重点专题。在“大咖沙发秀”环节,多位领域内大咖还对“ad危险因素及预防指南”、“ad治疗的前景与未来”、“如何做好ad全程管理”等话题进行精彩点评和热点讨论,干货满满、现场气氛强烈。

大会主席贾建平教授致辞(左)、卫材中国药业副总经理兼医药事业本部本部长张建忠在成都分会场致辞(右)
本次峰会线上线下互动热烈,各地区领域内的专家学者们汇聚一堂,共同收获最新的学术前沿进展与临床实践经验。

北京、上海、广州、成都四地分会场(线下)
国际阿尔茨海默病协会(adi)2019年统计数据显示,全球痴呆患者人数已突破5000万大关,到2050年预计患病人数将增至1.52亿人,每3秒就有一例新增患者。在中国,痴呆患者人数已超过1000万,患者平均存活年限仅5.9年。然而,尽管临床治疗需求巨大,但目前ad发病机制尚不明确,现有已上市的治疗药物大多仅能起到缓解症状的作用,迄今为止尚没有逆转或阻止病情进展的有效药物。而且,国际多个关于ad的药物临床试验均宣告失败,所以仍需要加大对ad治疗药物的研究与开发。
阿尔茨海默病是卫材长期关注的疾病领域,不仅将多奈哌齐引入中国并推动其进入医保,同时长期投入新药物的研发。卫材中国一直秉承hhc(human health care)“关心人类健康”的企业理念,将卫材在全球其它市场积累的丰富经验带入中国市场,在华发展的20年间,通过开展“记得我爱你”重新认识阿尔茨海默病公益行动、黄手环行动、记忆门诊、认知学院等科普活动及学术平台搭建,与各界联合,协力提升整个中国社会对阿尔茨海默病的关注与正确认识,造福广大患者及其家属。
卫材株式会社(总部:东京,首席执行官:内藤晴夫,“卫材”)宣布,在2020年欧洲内科肿瘤学会(esmo)虚拟大会上展示了内部发现的抗癌治疗halaven(海乐卫)(通用名:甲磺酸艾立布林,“艾立布林”)新脂质体制剂(e7389-lf)一期临床试验中her2阴性乳腺癌患者队列的最新结果(摘要编号:346p)。
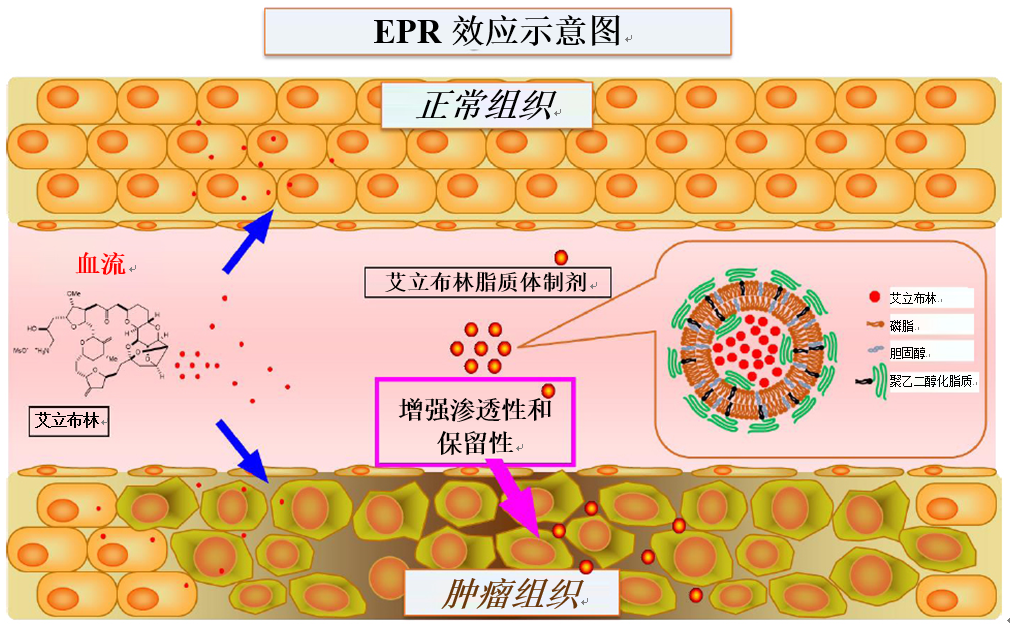
e7389-lf是一种新制剂,使用由脂质双层制成的脂质体来封装软海绵素类微管动力学抑制剂艾立布林。在肿瘤组织中,由于不完全的血管系统,血管内皮细胞之间存在视为允许大分子渗透的间隙。这种情况,除了不完全的淋巴功能之外,预计还能通过增强渗透性和保留性(epr)效应,使得能够分别在肿瘤中以比正常组织更大的量输送和保留包括脂质体制剂在内的高分子量药物。因此,e7389-lf预计可以提高肿瘤组织中艾立布林的浓度。
本次展示报告了入选28例复发性(her2阴性)乳腺癌患者(激素受体阳性:21例,三阴性:7例)的队列中e7389-lf的疗效和安全性结果,这些患者以前接受过蒽环类或紫杉醇类治疗,并且以前没有接受过艾立布林治疗(数据截止日期:2020年1月24日,无进展生存期和整体生存期截止日期:2020年4月17日),作为以前接受过治疗的选定实体瘤患者的开放标签一期临床试验(研究114)的一部分患者每三周静脉注射一次e7389-lf 2.0mg/m2体表面积(作为游离艾立布林),并且证明在全部her2阴性乳腺癌队列中,整体缓解率为35.7%(95%置信区间(ci):18.6-55.9)。在该队列中,证明了激素受体阳性患者的orr为42.9%(95%置信区间:21.8-66.6),三阴性患者的orr为14.3%(95%置信区间:0.4-57.9)。疾病控制率包括病情稳定率、部分缓解率和完全缓解率为89.3%(95%置信区间:71.8-97.7)。此外,中位无进展生存期(pfs)为5.7个月(95%置信区间:3.9-8.3),未达到中位整体生存期(os)(95%置信区间:10.3-未达到)。3级或以上(前5名)的不良事件为中性粒细胞减少症(67.9%)、白细胞减少症(42.9%)、血小板减少症(32.1%)、发热性中性粒细胞减少症(25.0%)和丙氨酸转氨酶升高(21.4%),这与迄今为止艾立布林的安全特征一致。此外,用g-csf(粒细胞集落刺激因子)乙二醇化非格司亭进行预防性治疗后,证明了发热性中性粒细胞减少症发生率降低(治疗组:10.0%,未治疗组:33.3%)。
卫材将肿瘤领域定位为一个关键的治疗领域,旨在发现具有治愈癌症潜力的革命性新药。卫材将继续在基于前沿癌症研究的新药开发方面创新,为进一步满足癌症患者及其家人和医疗保健提供者的多样化需求,并提高他们的福祉而做出努力。
媒体咨询:
卫材株式会社
公共关系部
81-(0)3-3817-5120
[编辑附注]
e7389-lf是一种新制剂,设计用于通过脂质体包裹更有效地将软海绵素类微管动力学抑制剂“halaven”(海乐卫)(甲磺酸艾立布林)输送到癌细胞中。目前正在日本进行一项关于选定实体肿瘤的一期临床研究。此外,目前正在日本与ono pharmaceutical co., ltd.合作进行一项关于靶向选定实体肿瘤的e7389-lf和纳武单抗联合疗法的ib/ii期临床试验
(经东京理科大学nishikawa教授许可后修改了图文)
halaven(海乐卫)属于软海绵素类微管动力学抑制剂,具有全新的作用机制。从结构上来说,halaven(海乐卫)是一种简化的和合成生产的软海绵素b,这是一种从海洋冈田软海绵中分离出来的天然产物。halaven(海乐卫)被认为是通过抑制微管动力学的生长阶段来起作用的,这样可防止细胞分裂。非临床研究还显示其对肿瘤微环境具有独特的作用,例如可增加肿瘤核心的血管灌注和通透性1、促进上皮状态改变,以及降低乳腺癌细胞的转移能力2等。
halaven(海乐卫)于2010年11月首次在美国获批用于转移性乳腺癌患者的治疗。halaven(海乐卫)目前获批准用于治疗全球超过75个国家的乳腺癌患者,包括日本、中国以及欧洲、美洲和亚洲国家。
此外,halaven(海乐卫)于2016年1月在美国首次获批准用于治疗软组织肉瘤,并且在包括日本、欧洲和亚洲在内的65个国家获得批准。此外,在美国和日本,halaven(海乐卫)已指定为用于软组织肉瘤的孤儿药。
具体而言,halaven(海乐卫)已获批用于以下适应症。
在美国,用于治疗如下患者:
在日本,用于治疗如下患者:
在欧洲,用于治疗如下成人患者:
在以前接受过蒽环类和紫杉类药物治疗的局部晚期或转移性乳腺癌患者中,对halaven(海乐卫)进行了两项开放标签、随机化、三期试验(embrace和301研究)。基于组合结果的汇总分析3,与对照组相比,halaven(海乐卫)组的整体生存期(os)显著延长(风险比为0.85[95%置信区间(ci)=0.77-0.95]p=0.003,halaven(海乐卫)中位os:15.2个月,对照组中位os:12.8个月)。与对照组相比,halaven(海乐卫)组的无进展生存期(pfs)延长(风险比为0.90[95%置信区间=0.81-0.997]p=0.046,halaven(海乐卫)中位pfs:4.0个月,对照组中位pfs:3.4个月)。
her2阴性乳腺癌组的整体缓解率orr)为13.5%。在该组中,激素受体阳性患者的orr为14.3%,三阴性患者的orr为12.0%。her2阴性乳腺癌组的中位pfs为4.0个月。
关于组合分析的安全性特征,各临床研究之间没有重大差异。这些三期研究中的患者每21天接受一次halaven(海乐卫)(第1天和第8天静脉注射1.4mg/m2)。
1 funahashi y et al., eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. cancer sci., 2014; 105, 1334-1342
2 yoshida t et al., eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (emt) to mesenchymal-epithelial transition (met) states. br j cancer, 2014; 110, 1497-1505
3 twelves c et al., efficacy of eribulin in women with metastatic breast cancer: a pooled analysis of two phase 3 studies. breast cancer res treat, 2014; 148, 553-561